当地时间2026年3月23日下午,欧洲血液与骨髓移植学会(EBMT)年会在西班牙马德里进入第二个全体会议(P02),“先天性代谢缺陷疾病的基因治疗:临床现况、剩余挑战及未来方向”专场,由意大利热那亚Emanuele Angelucci教授与西班牙马德里Antonio Perez Martinez教授联合主持。这场具有里程碑意义的会议汇聚了来自西班牙、意大利、德国、英国等多国的顶尖专家,聚焦范可尼贫血、溶酶体贮积症、血红蛋白病及原发性免疫缺陷四大疾病领域,系统梳理了基因治疗从实验室走向临床的征程。在全球首款CRISPR基因编辑药物获批、多款慢病毒基因治疗产品上市的背景下,本次会议不仅展示了技术突破的辉煌成果,更直面可及性、安全性与可持续性的现实挑战,为这一变革性疗法的未来发展指明方向。《血液时讯》特梳理专场精粹,以飨读者。
会议伊始,主席Antonio Perez Martinez教授在开场致辞中强调,医学界正处于关键转折点。长期以来,异基因干细胞移植是提高先天性代谢缺陷患者生存率的主要手段,但如今基因治疗已成为切实可选的替代方案。Emanuele Angelucci教授补充道,本次会议将呈现从实验室到临床的最新进展,四位讲者均为各自领域的先驱者。
01
范可尼贫血基因治疗:无预处理方案的突破
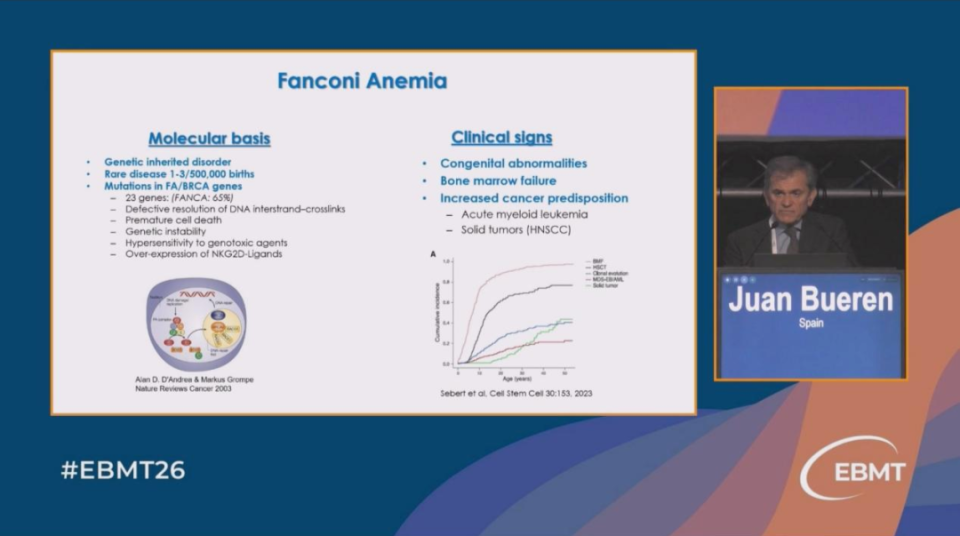
来自西班牙马德里的Juan Bueren教授分享了其团队二十余年深耕范可尼贫血(FA)基因治疗的突破性成果。异基因移植虽为唯一获批治愈手段,但面临高并发症风险及移植后癌症易感性增加的困境。Bueren教授指出,早期四项FA基因治疗临床试验均告失败,核心瓶颈在于干细胞的收集与基因校正效率。其团队通过临床前研究首次证明,FA患者基因校正后的造血干细胞具有显著增殖优势——这一发现为“无预处理方案”奠定了理论基础。
2016年启动的FANCOLEN-1试验采用创新策略:对FANCA型患者使用非格司亭联合普乐沙福动员CD34+细胞,经慢病毒载体体外转导后回输,完全跳过化疗预处理。首批4例患者数据显示,尽管植入缓慢,但所有患者外周血基因校正细胞比例逐渐增加。随访10年结果显示,回输高剂量CD34+细胞(≥40万/kg)的3例患者中,2例达到近100%基因校正,骨髓衰竭得到稳定甚至纠正。
与Rocket Pharmaceuticals合作的国际II期试验进一步验证了这一策略:在骨髓衰竭征兆出现前早期干预,CD34+细胞回输量增加三倍以上,植入速度显著加快。截至2025年3月,9例接受治疗剂量回输的患者中7例获得持续遗传学和表型校正。值得注意的是,II期试验中观察到的1例T细胞淋巴母细胞淋巴瘤经证实与基因治疗无关。
Bueren教授总结十年经验指出,CD34+细胞基数可预测动员效果,无预处理方案安全有效,阈值剂量的CD34+细胞回输可在大多数患者中介导临床和分子反应。展望未来,团队正拓展至FANCC和FANCG亚型,并探索先导编辑(prime editing,PE)及体内基因治疗新策略,同时开发针对FA患者头颈部鳞状细胞癌的CAR-T细胞疗法。
02
溶酶体贮积症:从“超正常酶工厂”到平台化开发
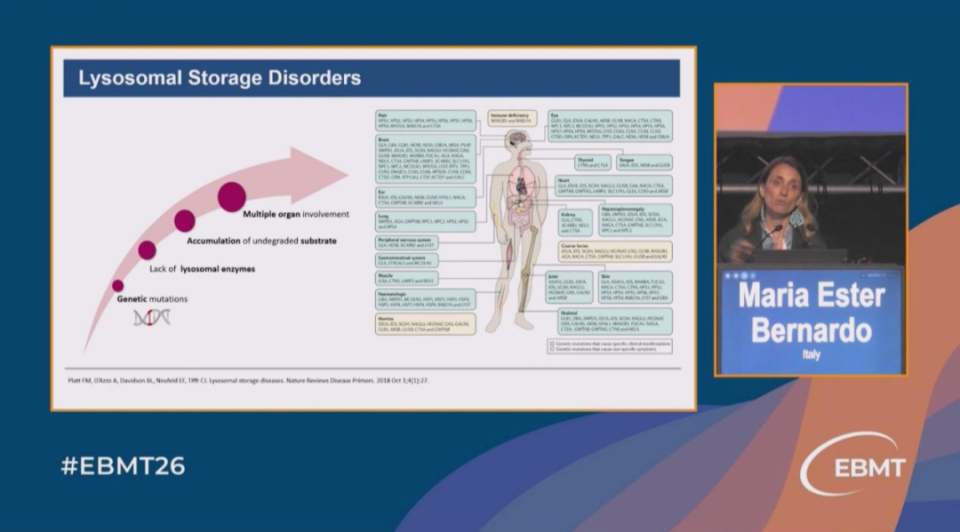
意大利米兰的Maria Ester Bernardo教授系统阐述了溶酶体贮积症(LSDs)基因治疗的临床现状与未来方向。LSDs是一组超过70种单基因遗传病,传统酶替代疗法和异基因移植均存在无法充分靶向中枢神经系统和骨骼系统的局限。基因治疗的核心理念是将自体造血干细胞转化为“超功能酶工厂”,通过超正常浓度的酶产生与分发,实现远离骨髓的器官(如大脑、骨骼)的交叉校正。
异染性脑白质营养不良(MLD)是首个成功应用慢病毒体外基因治疗的LSD。38例患者的中位随访近7年(最长12年)数据显示,基因修饰细胞高水平持续植入,脑脊液ARSA活性恢复正常,运动与认知功能显著改善,而自然病史患者则持续恶化。该疗法已获欧美上市许可,适用于无症状晚期婴儿型、早期青少年型及有症状早期青少年型患者。
Hurler综合征(黏多糖贮积症I型最严重形式)的治疗更具挑战性,涉及神经系统、骨骼系统、心肌病等多系统受累。8例无匹配供体患儿接受IDUA基因慢病毒载体治疗后,5年随访显示持续植入、酶水平超正常、尿液毒性物质清除良好,认知运动技能逐渐获得,纵向生长符合WHO标准,髋臼发育不良及脊柱改变参数稳定或改善。一项比较基因治疗与异基因移植的III期随机试验正在进行中。
Bernardo教授还介绍了MPS IIIA(SGSH缺陷)和MPS II(Hurler综合征)的积极临床数据,以及肾上腺脑白质营养不良(ALD)的积极长期随访结果。同时,她警示了相关安全性信号:两项试验中67例ALD患者出现7例血液系统恶性肿瘤(MDS/AML),分析证实与MECOM原癌基因附近的转基因插入存在因果关系,可能与使用强病毒启动子MNDU相关。
面对商业可持续性挑战,Bernardo教授提出平台化开发策略:基于MLD和MPS I的经验,针对MPS IV A、GLB1相关疾病、α-甘露糖苷贮积症等选定LSDs并行开发,采用主方案(master protocol)设计,以更快、更低成本推进临床试验,实现“即插即用”的广泛适用系统。她强调,新生儿筛查的早期诊断、真实世界数据的长期监测,以及针对无效突变患者的免疫抑制策略,是未来亟需解决的关键问题。
03
血红蛋白病基因治疗:CRISPR时代的机遇与反思

德国Selim Corbacioglu教授聚焦血红蛋白病(镰状细胞病与地中海贫血)的基因编辑治疗,指出这一领域面临独特的规模化挑战——每年全球新增20-50万患者,绝大多数居住在低收入和中等收入国家。尽管常规治疗取得一定进展,但患者生存期仍显著缩短,治愈需求迫切。
当前已上市产品包括Bluebird Bio的基因添加疗法Zynteglo和Vertex的CRISPR-Cas9基因编辑疗法Casgevy。Casgevy通过敲除BCL11A解除对γ-链转录的抑制,上调胎儿血红蛋白(HbF)产生。CLIMB-121/131试验显示,镰状细胞病患者血管闭塞危象几乎完全消除;CLIMB-111试验中,成年及青少年地中海贫血患者完全摆脱输血依赖。
然而,Corbacioglu教授也尖锐地指出了“基因编辑1.0版”的诸多局限:溶血持续存在(约60%血红蛋白S残留)、干细胞采集困难(约20%失败率,需多次单采)、白消安预处理的严重毒性(静脉闭塞病、黏膜炎、可逆性后部脑病综合征风险)、植入延迟(中性粒细胞最长56天,血小板最长200天)。
Beam Therapeutics推出的“基因编辑2.0版”碱基编辑(BEACON研究)带来了新希望:采用腺苷碱基编辑HBG1/2启动子(无双链断裂),单采循环减少至1-3次,植入时间缩短至单倍体移植水平,HbF比例更高且溶血标志物正常化。但早期数据中仍出现1例白消安相关肺部并发症死亡,凸显预处理优化的紧迫性。
真实世界数据则揭示了严峻的可及性鸿沟:即使在高收入国家,仅15%-20%镰状细胞病患者符合当前适应症(年龄限制、排除合并症等),单采失败率与供体可用性进一步缩小适用人群。Corbacioglu教授呼吁建立非恶性疾病商业化基因治疗真实世界数据平台,解决新适应症扩展与长期安全性监测问题。他强调,白消安应被更安全药物替代,而学术界需推动不依赖商业利益的前瞻性试验,从而实现治疗公平。
04
原发性免疫缺陷:从γ-逆转录病毒到体内基因治疗

英国伦敦的Claire Booth教授以向基因治疗先驱致敬开场——30年前首位接受ADA-SCID T细胞基因治疗的Ashanti DeSilva和首位接受γ-逆转录病毒治疗X-SCID的Rhys目前均状况良好。她指出,先天性免疫缺陷(IEI)是基因治疗的“基础测试场”,异基因移植曾是超过500种疾病的标准治疗,而基因治疗通过避免同种异体反应、降低预处理强度、免除免疫抑制,提供了毒性更小的替代方案。近年来,多维技术改进显著提升了临床结果,包括非格司亭联合普乐沙福动员外周血细胞、慢病毒载体替代γ-逆转录病毒、产品冷冻保存确保质量、优化植入等。
Booth教授分享了X-SCID慢病毒基因治疗多中心试验的未发表数据:20例患者招募完成,中位随访3.5年,17例可评估患者中16例达到主要终点,14例停止免疫球蛋白替代并对疫苗产生反应,T细胞恢复迅速且优于异基因移植和早期试验,多谱系基因标记持续稳定。其他成功范例包括:RAG-1 SCID跨国多中心试验、X-CGD与Wiskott-Aldrich综合征的长期安全性数据、ADA-SCID 62例患者近500患者年随访等。LAD-I相关试验中,采用乌司奴单抗抑制炎症负担,患者感染与住院显著减少,腹部病变完全消退,该疗法正接受FDA上市审批。
针对主要累及T细胞的疾病,自体基因修饰T细胞策略展现出独特优势:无需动员、高转导效率、低基因毒性风险、毒性较小的淋巴细胞清除预处理。IPEX综合征(Treg细胞病)慢病毒T细胞基因治疗已治疗10例患者,结果值得期待;X-连锁淋巴增生症(XLP)T细胞试验将于年内启动。基于HDR的基因编辑T细胞也已进入临床:针对CD40配体缺乏症的CD4 T细胞编辑,以及针对CTLA-4单倍剂量不足的T细胞编辑,均显示出持久性与靶向归巢能力。
Booth教授强调,体内基因治疗被视为“改变游戏规则”的下一代技术。Ensoma赞助的X-CGD试验中,首位患者已接受病毒样颗粒(VLP)递送CYBB基因的体内HSC靶向治疗,配合MGMT选择基因和睡美人转座酶可实现持久校正,首批结果预计年中公布。针对p47-CGD热点突变的引导编辑试验也取得了高水平编辑与中性粒细胞校正,技术可行性已获验证。此外,非营利模式对罕见病治疗的可持续至关重要。美国FDA的“可信机制(plausible mechanism)”路径与英国药品和健康产品管理局(MHRA)罕见病新许可策略等监管创新,有望为平台化审批带来曙光。
05
深度讨论:可及性、可持续性与科学责任
在讨论环节,与会者围绕基因治疗的现实挑战展开深度交锋。关于预处理的优化,Corbacioglu教授回应指出,Grafapex(treosulfan,曲奥舒凡)不穿过血脑屏障、不经肝脏代谢、炎症负担更轻,有望改善植入微环境,并呼吁积极生成临床证据以推动替代白消安。
面对单倍体移植技术进步的提问,Booth教授指出,基因治疗具有无GVHD风险、低毒性预处理、无需免疫抑制的固有优势,使其在具有合并症的患者中更具吸引力,但可及性与成本仍是关键变量。
谈到全球可及性与经济成本时,多位教授共同呼吁,当前“口袋里装满奇妙治疗却无法带给患者”的现状不可持续。Corbacioglu教授强调学术界需推动不依赖市场价值的前瞻性试验;Bueren教授补充指出,需开发更简单、低成本的非病毒载体,监管机构应接受合理风险以降低成本;Booth教授提出基于学术网络、即时制造(point-of-care)和非营利模型的替代经济路径。
总结
本次全体会议展现了当前先天性代谢缺陷基因治疗的临床状态与未竟征程。从技术维度看,慢病毒载体已证明长期安全性与有效性,CRISPR-Cas9碱基编辑开启精准调控新纪元,体内基因治疗与引导编辑预示“去体外化”的未来;从疾病维度看,免疫缺陷的多策略覆盖,共同勾勒出基因治疗从“挽救生命”到“治愈生命”的进化轨迹。然而,讨论环节揭示的“可及性鸿沟”正呼唤监管创新、非营利模式与全球协作的系统性变革。未来的基因治疗,不仅需要更精妙的治疗工具,更需要更包容的价值共识——让每一个生命,无论地理与经济境遇,都能平等地享有治愈的希望。